Patchwork
Welcome
Patchwork is a bioinformatic tool for analyzing and visualizing allele-specific copy numbers and loss-of-heterozygosity in cancer genomes. The data input is in the format of whole-genome sequencing data which enables characterization of genomic alterations ranging in size from point mutations to entire chromosomes. High quality results are obtained even if samples have low coverage, ~4x, low tumor cell content or are aneuploid.
Patchwork is available for two data types. Patchwork takes BAM files as input whereas PatchworkCG takes input from CompleteGenomics files.
TAPS performs the same analysis as Patchwork but for microarray data.
Detailed guides and information regarding all three of these bioinformatic tools can be found in their respective tabs.
If you have any feedback or questions please do not hesitate to contact us!
Publications
Patchwork: allele-specific copy number analysis of whole genome sequenced tumor tissueMarkus Mayrhofer, Sebastian DiLorenzo and Anders Isaksson
Genome Biology 2013, 14:R24 doi:10.1186/gb-2013-14-3-r24
Published: 25 March 2013
Allele-specific copy number analysis of tumor samples with aneuploidy and tumor heterogeneity.
Rasmussen M, Sundström M, Göransson Kultima H, Botling J, Micke P, Birgisson H, Glimelius B, Isaksson A.
Genome Biology 2011 Oct 12:R108. doi: 10.1186/gb-2011-12-10-r108.
Published: 24 October 2011

Begin by starting R. It is recommended that you use the latest version.
Issue the following commands within R to install patchworkCG.
install.packages("patchworkCG", repos="http://R-Forge.R-project.org")
If for some reason that does not work add the 'type="source"' to it, as so:
install.packages("patchworkCG", repos="http://R-Forge.R-project.org",type="source")
If something still does not work with installation, here is a link to the location of the source files: Patchwork
Specifically the files ASM/masterVarBeta, CNV/depthOfCoverage and CNV/somaticCnvSegmentsNondiploidBeta are used.

patchwork.CG.plot()
patchwork.CG.plot() is a function that visualizes allele-specific copy numbers from the whole-genome sequenced data of tumors.It is recommended that you run patchworkCG from a "clean" working directory. This prevents the risk of having files write over eachother when you run multiple different samples.
In the R environment load the patchworkCG library:
library(patchworkCG)Read the excellent documentation for patchwork.CG.plot():
?patchwork.CG.plotExcerpt from ?patchwork.CG.plot regarding usage and arguments:
Usage:
patchwork.CG.plot(path,name='CG_sample',manual_file_input=FALSE,masterVarBeta=NULL,somaticCnvSegments=NULL,
depthOfCoverage=NULL)
Arguments:
path: Path to the ASM folder. One of the subfolders of your
completegenomics directory.
name: Default is 'CG_sample'. The name you wish associated with
the plots that will be generated.
manual_file_input: Default is FALSE. If you set it to TRUE you will be
prompted to provide path and filename for the masterVarBeta,
somaticCnvSegmentsNondiploid and depthOfCoverage file.
masterVarBeta: Path to and COMPLETE NAME of the masterVarBeta file,
should you wish to implement it directly. Useful if you want
to run the program on multiple samples and wish to create a
script going from file to file. This saves you from having to
conserve the CompleteGenomics file structure. Default is
NULL.
somaticCnvSegments: Path to and COMPLETE NAME of the somaticCnvSegments
file, should you wish to implement it directly. Useful if you
want to run the program on multiple samples and wish to
create a script going from file to file. This saves you from
having to conserve the CompleteGenomics file structure.
Default is NULL.
depthOfCoverage: Path to and COMPLETE NAME of the depthOfCoverage file,
should you wish to implement it directly. Useful if you want
to run the program on multiple samples and wish to create a
script going from file to file. This saves you from having to
conserve the CompleteGenomics file structure. Default is
NULL.
Example run of patchwork.CG.plot() where output has been left for display: patchwork.CG.plot(path="path/to/ASM/") Reading files from ASM folder File input complete Performing calculations Calculations complete Saving objects to CG.Rdata Initiating Plotting Plotting Complete patchwork.CG.plot Complete Please read documentation on running patchwork.CG.copynumbersOnce it is complete there should be several plots in your working directory as well as the file "CG.Rdata".
Execution should take no longer than half an hour, depending on your system.
patchwork.CG.copynumbers()
patchwork.CG.copynumbers() is a function that determines which relationship between coverage and allelic imbalance signifies which copy number and allele ratio for each segment.Running patchwork.CG.copynumbers() requires the aforementioned CG.Rdata file, the function will find it automatically if it is in your working directory. There is also the option to point to the file using the "CGfile" argument of the function.
Read the documentation for patchwork.CG.copynumbers():
?patchwork.CG.copynumbersExcerpt from ?patchwork.CG.copynumbers regarding usage and arguments:
Usage:
patchwork.CG.copynumbers(cn2,delta,het,hom,maxCn=8,ceiling=1,name="copynumbers_",
CGfile=NULL,forcedelta=F)
Arguments:
cn2: The approximate position of copy number 2, diploid, on total
intensity axis.
delta: The difference in total intensity between consecutive copy
numbers. For example 1 and 2 or 2 and 3. If copy number 2
has total intensity ~0.6 and copy number 3 har total
intensity ~0.8 then delta would be 0.2.
het: Allelic imbalance ratio of heterozygous copy number 2.
hom: Allelic imbalance ratio of Loss-of-Heterozygosity copy number
2.
maxCn: Highest copy number to calculate for. Default is 8.
ceiling: Default is 1.
name: Default is "copynumbers_". The name you want attached to generated
plots.
CGfile: Default is NULL. If your CG.Rdata file is not in your working
directory, and you dont wish to move it to your working
directory, you can simply input the path here as CGfile =
"path/to/file/CG.Rdata" so patchwork.CG.copynumbers() can find its
data.
forcedelta: Default is FALSE. If TRUE the delta value will be absolute
and not subject to adjustments.
To infer the arguments for patchwork.CG.copynumbers() you will need to look at one of the chromosomal
plots generated using patchwork.CG.plot(). Here is the topmost section of one such plot with some added bars
and text for tutorial purposes. The plot shows the whole genome in grey and the chosen chromosome, not
important in this guide, in red to blue gradient. From this whole genome picture you can then use the
arrangement of the clustered areas to estimate copy numbers and allele-specific information. 
The cn2 argument is the position of copy number 2. In this example cn2 is ~0.8.
The delta argument is the difference between two copynumbers on the coverage axis. In this example we used copy number 2 and copy number 3, but we could as well have used copy number 2 and the unmarked copy number 1 to the left of copy number 2. Delta does not take negative argument input. In this example delta is ~0.28.
The het argument is the position of heterozygous copy number 2 on the allelic imbalance axis. In this example het is ~0.21.
The hom argument is the position of homozygous, loss-of-heterozygosity, copy number 2 on the allelic imbalance axis. In this example hom is ~0.79.
Usually you do not need to bother with maxCn and ceiling arguments.
There is more information on interpreting the plots in the results tab which may aid you in determining the parameters.
An example run of patchwork.CG.copynumbers():
patchwork.CG.copynumbers(cn2=0.8, delta=0.28, het=0.21, hom=0.79, CGfile="path/to/CG.Rdata")This will generate an additional set of plots in your working directory.
Plots of patchwork.CG.plot()
Here is an example plot of chromosome 1, 70% tumor cells, generated from HCC1187.After running patchwork.CG.plot() you should have 24 of these in your working directory, one for each chromosome.
For information regarding allelic imbalance and coverage click here.
Top part of the plot
The clusters in the plot display regions of certain allelic constitution and copy number. The copy number increases along the Coverage axis while paternal/maternal allele ratio becomes less balanced along the Allelic Imbalance axis. We can use this information to determine the clusters probable copy number and allele content.The chromosome in question is colored against a background of the complete genome in grey. A colored circles gradient and size correlate with its segments position and size on the chromosome. Colors of segments match in regard to all three parts of the plot. The circles are semi-transparent so a darker hue, both for colored and grey, indicate greater amount of genomic content in that region.
We know that each cluster has a certain copy number and allele content and we know that the average copy number of the genome in question is at position 1 on the Coverage axis. The far left cluster is the deletions, copy number 1. After copy number 1 we see two clusters, albeit the lower one does not have much content. They are most likely the two allelic states of copy number 2. Continuing with this reasoning the next set of clusters is copy number 3, etc. This arrangement of the genome is easier to see from one of the plots generated by patchwork.CG.copynumbers(), or in our section above on allelic imbalance, however before using that function you will need to be able to determine the constitution of the tumor genome as it is used as input arguments for patchwork.CG.copynumbers()!

Middle part of the plot
The chromosome in questions coverage plotted against the position on the chromosome. Note how the segments colors match the Top circles and Bottom segments.Bottom part of the plot
The chromosome in questions allelic imbalance plotted against the position on the chromosome.Plots of patchwork.CG.copynumbers()
This plot, arbitrarily named "check", is not chromosome specific and shows the complete tumor genome with assigned tags for copy number and allele content, much like the explanatory picture seen previously but with text describing the copy numbers and allele ratios of the segments rather than purple and green bars.
1m0 is copy number 1, homozygous.
2m1 is copy number 2, heterozygous.
2m0 is copy number 2, homozygous.
3m1 is copy number 3, heterozygous.
3m0 is copy number 3, homozygous.
And so on.
The other chromosomal plots generated from patchwork.CG.copynumbers() are the same as the ones generated by patchwork.CG.plot() but with one extra part of the plot.

In the part of the plot seen second from the top we see the total copy number, colored, and minor copy number, grey, of a segment. If a segments total copy number is 4 and minor copy number is 2 then that segment of the chromosome has 2 paternal and 2 maternal copies.
Another example; if a segments total copy number is 3 and minor copy number is 0 then that segment of the chromosome has 3 paternal OR 3 maternal copies.
It builds upon the knowledge gained from the other tabs, installation to results, and as such is not overly detailed. Read the other tabs before doing the demo!
Used in this demo is the publically available cancer data set HCC2218 of CompleteGenomics.
We will go through the exact commands applicable in a typical run of this data and some plots that you can compare your own results with.
Once you have downloaded the data and unpacked it you should have a file structure resembling the screenshot below. The most important files, which are used in patchworkCG, have been highlighted.

Now we are ready to start R and issue all the necessary commands, as seen in the install and execution tabs.
#For a working directory I chose just above the ASM folder.
$ GS00258-DNA_B03 ls
ASM idMap-HCC2218-H-200-37-ASM-T1.tsv
HCC2218-H-200-37-ASM version
LIB
#Starting R
$ GS00258-DNA_B03 R
R version 2.14.1 (2011-12-22)
Copyright (C) 2011 The R Foundation for Statistical Computing
ISBN 3-900051-07-0
Platform: x86_64-apple-darwin9.8.0/x86_64 (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
Natural language support but running in an English locale
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
#Install package
> install.packages("patchworkCG", repos="http://R-Forge.R-project.org",type="source")
trying URL 'http://R-Forge.R-project.org/src/contrib/patchworkCG_1.0.tar.gz'
Content type 'application/x-gzip' length 669849 bytes (654 Kb)
opened URL
==================================================
downloaded 654 Kb
* installing *source* package 'patchworkCG' ...
** R
** data
** preparing package for lazy loading
** help
*** installing help indices
** building package indices ...
** testing if installed package can be loaded
* DONE (patchworkCG)
The downloaded packages are in
'/private/var/folders/zm/8v842d8n4172k1yc0589p8sw0000gn/T/RtmprYh5Mq/downloaded_packages'
#Load Library
> library(patchworkCG)
#Execute patchwork.CG.plot
> patchwork.CG.plot(path="ASM/",name="Demo_")
Reading files from ASM folder
File input complete
Performing calculations
Calculations complete
Saving objects to CG.Rdata
Initiating Plotting
Plotting Complete
patchwork.CG.plot Complete
Please read documentation on running patchwork.CG.copynumbers
#Execute patchwork.CG.copynumbers (See execution section for aquiring arguments)
#Side note: this is a tricky sample as there is no copy number 2 with loss
#of heterozygosity, but as copynumber 1 has allelic imbalance around 1 and copy number 3
#also has high allelic imbalance we can estimate that had it existed copy number 2
#would have had very high allelic imbalance. So we approximate it to 0.96.
> patchwork.CG.copynumbers(cn2=0.9,delta=0.5,het=0.25,hom=0.96)
Warning message:
NAs introduced by coercion
#Quit
> q()
Save workspace image? [y/n/c]: n
$ GS00258-DNA_B03 ls
ASM Demo__KaCh_chr1.png
CG.Rdata Demo__KaCh_chr10.png
copynumbers.Rdata Demo__KaCh_chr11.png
copynumbers_KaChCN_chr1.png Demo__KaCh_chr12.png
copynumbers_KaChCN_chr10.png Demo__KaCh_chr13.png
copynumbers_KaChCN_chr11.png Demo__KaCh_chr14.png
copynumbers_KaChCN_chr12.png Demo__KaCh_chr15.png
copynumbers_KaChCN_chr13.png Demo__KaCh_chr16.png
copynumbers_KaChCN_chr14.png Demo__KaCh_chr17.png
copynumbers_KaChCN_chr15.png Demo__KaCh_chr18.png
copynumbers_KaChCN_chr16.png Demo__KaCh_chr19.png
copynumbers_KaChCN_chr17.png Demo__KaCh_chr2.png
copynumbers_KaChCN_chr18.png Demo__KaCh_chr20.png
copynumbers_KaChCN_chr19.png Demo__KaCh_chr21.png
copynumbers_KaChCN_chr2.png Demo__KaCh_chr22.png
copynumbers_KaChCN_chr20.png Demo__KaCh_chr3.png
copynumbers_KaChCN_chr21.png Demo__KaCh_chr4.png
copynumbers_KaChCN_chr22.png Demo__KaCh_chr5.png
copynumbers_KaChCN_chr3.png Demo__KaCh_chr6.png
copynumbers_KaChCN_chr4.png Demo__KaCh_chr7.png
copynumbers_KaChCN_chr5.png Demo__KaCh_chr8.png
copynumbers_KaChCN_chr6.png Demo__KaCh_chr9.png
copynumbers_KaChCN_chr7.png Demo__KaCh_chrX.png
copynumbers_KaChCN_chr8.png Demo__KaCh_chrY.png
copynumbers_KaChCN_chr9.png Demo__karyotype.png
copynumbers_KaChCN_chrX.png HCC2218-H-200-37-ASM
copynumbers_KaChCN_chrY.png LIB
copynumbers_Ka_check.png idMap-HCC2218-H-200-37-ASM-T1.tsv
Copynumbers.csv version
If successfull your working directory should now contain:
- 50 plots
- CG.Rdata
- copynumbers.Rdata
- Copynumbers.csv
Here are three of the plots generated during the demo run to validate your plots with.
Demo__KaCh_chr1.png
copynumbers_KaChCN_chr1.png
copynumbers_Ka_check.png
Please feel free to contact us with any questions, suggestions or feedback!
Begin by starting R. It is recommended that you use the latest version.
To install DNACopy, which is a program patchwork needs to run, issue the following commands within R.
source("http://bioconductor.org/biocLite.R")
biocLite("DNAcopy")
After DNAcopy is installed you can install patchwork and patchworkData.
install.packages("patchworkData", repos="http://R-Forge.R-project.org")
install.packages("patchwork", repos="http://R-Forge.R-project.org")
If for some reason that does not work add the 'type="source"' to it, as so:
install.packages("patchworkData", repos="http://R-Forge.R-project.org",type="source")
install.packages("patchwork", repos="http://R-Forge.R-project.org",type="source")
If something still does not work with installation, here are links to the location of the source files: Patchwork
DNACopy
Python and Pysam
Currently Pysam 0.7.4 and Python 2.6.7 and 2.7.1 are tested and supported.
To successfully run patchwork you will need to obtain/create these items:
- An aligned and sorted tumor BAM file.
A BAI index of your BAM file.
A pileup of your BAM file.
A VCF file of your tumor pileup (if applicable). - (optional) A matched normal sample to your tumor in BAM format.
A BAI index of your normal sample BAM.
A pileup of your normal sample BAM.
A VCF file of your normal pileup (if applicable). - (optional) A standard Reference file. (Illumina/Solexa, SOLiD or your own)
If you have a matched normal sample you should create a pileup and BAI file. The BAI file is required for the file to be read by patchwork.
You can also use a reference file, see below, if you do not have a matched normal.
The optional VCF files need to be supplied if you are using a later version of samtools and have opted for using samtools mpileup to create the pileup file. The vcfs contain consensus and variant information which the mpileup generated pileup lacks. We recommend using the pileup from older version of samtools rather than mpileup + vcf of new version. More information on generating these files is further down.
Here are two standard reference files created for patchwork for samples where alignment used the UCSC HG19 reference. It is very important that you choose a reference which matches the sequencing technology and reference genome used for your tumor sample.
Solexa/Illumina reference
SOLiD reference
If you wish to create your own reference file, for example one that works for HG18, use patchworks.createreference().
patchwork.createreference()
This function creates a reference using a pool of samples of your selection. Choosing the samples to use for reference creation you should consider these factors:
- Aligned using same reference genome (HG18/HG19)
- Sequenced using the same technique
- From the same organism
- Non-tumorous
- Same gender
To execute patchwork.createreference() start R and load the patchwork and patchworkData libraries:
library(patchwork) library(patchworkData)Read the documentation for patchwork.createreference():
?patchwork.createreferenceThe function takes a series of BAM files as input and outputs a single reference file. It also has a "output" parameter for customizing output name.
Execute the function, pointing to your desired files:
patchwork.createreference("file1","path/to/file2","../file3","~/here_is/file4",output="REFOUT")
This will generate REFOUT.Rdata, or whichever prefix you chose, in your working directory.
Use this file for the reference parameter of patchwork.plot(). SAMtools file preparation
To sort your BAM file:
samtools sort <tumorfile>.bam <sortedtumorfile>.bamTo create an index, BAI file, of either your normal sample or tumor sample BAM files:
samtools index <tumor_or_normalfile>.bamThe BAI file should always have the same name as your tumor file, for example "tumor.bam" would have "tumor.bam.bai". If patchwork.plot() cannot find a correct BAI file this error is displayed.
Instructions for SAMtools <= 0.1.16
samtools pileup -vcf <humangenome>.fasta <tumor_or_normalfile>.bam > pileupYour pileup file should have this format:
$less pileup chr1 10179 c W 0 0 60 5 ,,tna ##6!3 chr1 10180 t W 6 6 60 5 ,,,aa ##369 chr1 10377 a R 0 1 60 1 g @ chr1 11391 t A 0 3 60 1 a B chr1 18592 C Y 0 3 60 1 t B chr1 23359 c S 0 2 60 1 g A chr1 24067 A R 0 2 60 1 G A chr1 30315 G C 3 24 60 2 cC =; chr1 92200 a W 0 3 60 1 t B chr1 96592 T C 0 3 60 1 C B chr1 100140 a M 0 1 60 1 C @ chr1 104697 g K 0 2 60 1 T A chr1 127285 A R 0 3 60 1 g B
Instructions for SAMtools > 0.1.16
samtools mpileup -f <humangenome>.fasta <tumor_or_normal>.bam > mpileupFor consensus calling:
samtools mpileup -uf <humangenome>.fasta <tumor_or_normal>.bam | bcftools view -bvcg - > <unfiltered_output>.bcf bcftools view <unfiltered_output>.bcf | vcfutils.pl varFilter -D100 > <output>.vcfThe -D100 option filters out SNPs with a read depth higher than 100 (They might be from repeat regions in your sample), you should change this parameter based on the average coverage of your sample.
For average coverage 30x =~ -D100, average coverage 120x =~ -D500 etc.
Remember to use both of these files when running patchwork.plot() with the vcf file in the Tumor.vcf/Normal.vcf parameter and the mpileup in Tumor.pileup/Normal.pileup parameter.
You should now have all the components needed for execution!

patchwork.plot()
It is recommended that you run patchwork from a "clean" working directory to avoid the risk of overwriting when running patchwork on multiple samples.
Execution may take quite a while depending on the size of your sample, if possible, run it on a dedicated computer. (A 50GB tumor sample with a matched 50GB normal sample took ~6 hours and used 10GB (out of 24GB available) RAM. However there are many factors that influence runtime so be generous with time/RAM allocation!)
Start by initiating the R environment and loading the patchwork and patchworkData libraries:
library(patchwork) library(patchworkData)Read the documentation for patchwork.plot():
?patchwork.plotExcerpt from ?patchwork.plot regarding usage and arguments:
Usage:
patchwork.plot(Tumor.bam,Tumor.pileup,Tumor.vcf=NULL,Normal.bam=NULL,Normal.pileup=NULL,Normal.vcf=NULL,
Reference=NULL,Alpha=0.0001,SD=1)
Arguments:
Tumor.bam: Path to an aligned and sorted Tumor Bam-file.
Tumor.pileup: Pileup file generated by either
(SAMtools v 0.1.16 or older)
samtools -vcf reference.fasta tumor.bam > outfile
or (SAMtools v 0.1.17 or newer)
samtools mpileup -f reference.fasta tumor.bam > outfile
Tumor.vcf: Default is NULL. If samtools mpileup command has been used
you will need to generate a vcf file using
samtools mpileup -uf reference.fasta tumor.bam | bcftools
view -bvcg - > raw.bcf
and then
bcftools view raw.bcf | vcfutils.pl varFilter -D100 >
outfile.vcf
Where the -D100 option filters out SNPs with a read depth
higher than 100. It should be changed depending on coverage,
at 30x -D100 should be ok and at 120x -D500 might be more
suitable for example.
Normal.bam: Default is NULL. The matched normal sample of the your
Tumor.bam.
Normal.pileup: Default is NULL. The pileup of your normal sample
generated through
(SAMtools v 0.1.16 or older)
samtools -vcf reference.fasta normal.bam > outfile
or (SAMtools v 0.1.17 or newer)
samtools mpileup -f reference.fasta normal.bam > outfile
Normal.vcf: Default is NULL. If samtools mpileup command has been used
you will need to generate a vcf file using
samtools mpileup -uf reference.fasta normal.bam | bcftools view -vcg - > outfile.vcf
Reference: Default is NULL. Path to a reference file that can be
created using patchwork.createreference() or downloaded from
patchworks homepage when applicable.
Alpha: Default 0.0001, change if you want to try to get a better
segmentation from patchwork.segment(). From DNAcopy
(?segment): alpha: significance levels for the test to accept
change-points.
SD: Default 1, change if you want to try to get a better
segmentation from patchwork.segment(). From DNAcopy
(?segment): undo.SD: the number of SDs between means to keep
a split if undo.splits="sdundo".
Example run of patchwork.plot() where output has been left for display: patchwork.plot(Tumor.bam="patchwork.example.bam",Tumor.pileup="patchwork.example.pileup",Reference="../HCC1954/datasolexa.RData") Initiating Allele Data Generation Initiating Read Chromosomal Coverage Reading chr1 Reading chr2 Reading chr3 Reading chr4 Reading chr5 Reading chr6 Reading chr7 Reading chr8 Reading chr9 Reading chrX Reading chrY Reading chr10 Reading chr11 Reading chr12 Reading chr13 Reading chr14 Reading chr15 Reading chr16 Reading chr17 Reading chr18 Reading chr19 Reading chr20 Reading chr21 Reading chr22 Read Chromosomal Coverage Complete Initiating GC Content Normalization GC Content Normalization Complete Initiating Smoothing Smoothing Chromosome: chr1 Smoothing Chromosome: chr2 Smoothing Chromosome: chr3 Smoothing Chromosome: chr4 Smoothing Chromosome: chr5 Smoothing Chromosome: chr6 Smoothing Chromosome: chr7 Smoothing Chromosome: chr8 Smoothing Chromosome: chr9 Smoothing Chromosome: chrX Smoothing Chromosome: chrY Smoothing Chromosome: chr10 Smoothing Chromosome: chr11 Smoothing Chromosome: chr12 Smoothing Chromosome: chr13 Smoothing Chromosome: chr14 Smoothing Chromosome: chr15 Smoothing Chromosome: chr16 Smoothing Chromosome: chr17 Smoothing Chromosome: chr18 Smoothing Chromosome: chr19 Smoothing Chromosome: chr20 Smoothing Chromosome: chr21 Smoothing Chromosome: chr22 Smoothing Complete Initiating Segmentation Note: If segmentation fails to initiate the probable reason is that you have not installed the R package DNAcopy. See patchworks README for installation instructions. Analyzing: chr1.p Analyzing: chr1.q Analyzing: chr2.p Analyzing: chr2.q Analyzing: chr3.p Analyzing: chr3.q Analyzing: chr4.p Analyzing: chr4.q Analyzing: chr5.p Analyzing: chr5.q Analyzing: chr6.p Analyzing: chr6.q Analyzing: chr7.p Analyzing: chr7.q Analyzing: chr8.p Analyzing: chr8.q Analyzing: chr9.p Analyzing: chr9.q Analyzing: chrX.p Analyzing: chrX.q Analyzing: chrY.p Analyzing: chrY.q Analyzing: chr10.p Analyzing: chr10.q Analyzing: chr11.p Analyzing: chr11.q Analyzing: chr12.p Analyzing: chr12.q Analyzing: chr13.q Analyzing: chr14.q Analyzing: chr15.q Analyzing: chr16.p Analyzing: chr16.q Analyzing: chr17.p Analyzing: chr17.q Analyzing: chr18.p Analyzing: chr18.q Analyzing: chr19.p Analyzing: chr19.q Analyzing: chr20.p Analyzing: chr20.q Analyzing: chr21.p Analyzing: chr21.q Analyzing: chr22.q Segmentation Complete Initiating Segment data extraction (Medians and AI) Segment data extraction Complete Saving information objects needed for patchwork.copynumbers in (prefix)_copynumbers.Rdata Initiating Plotting Plotting Complete Shutting down..... Warning messages: (Some warning messages about trying to load the files in the checklist below is not uncommon)If you get any errors not mentioned on the homepage please send us an email and we will get back to you as soon as possible!
If execution went well your working directory should have 1 overview plot and 24 chromosomal plots.
The working directory should also contain these files, where (prefix) is the name of your BAM file without the .bam file ending:
- (prefix)_copynumbers.Rdata
- (prefix)_data.Rdata
- (prefix)_pile.alleles.Rdata
- (prefix)_Segments.Rdata
- (prefix)_smoothed.Rdata
- pile.alleles
patchwork.copynumbers()
The only file you absolutely must have in your working for the next part of execution is (prefix)_copynumbers.Rdata.
Read the documentation for patchwork.copynumbers():
?patchwork.copynumbersExcerpt from ?patchwork.copynumbers regarding usage and arguments:
Usage: patchwork.copynumbers(CNfile,cn2,delta,het,hom,maxCn=8,ceiling=1,forcedelta=F,male.sample=F,male2femref=F) Arguments: CNfile: The name and path of your copynumbers file, generated from patchwork.plot(). Example Myfile_copynumbers.Rdata. cn2: The approximate position of copy number 2,diploid, on total intensity / coverage axis. delta: The difference in total intensity between consecutive copy numbers. For example 1 and 2 or 2 and 3. If copy number 2 has total intensity ~0.6 and copy number 3 har total intensity ~0.8 then delta would be 0.2. het: Allelic imbalance ratio of heterozygous copy number 2. hom: Allelic imbalance ratio of Loss-of-heterozygosity copy number 2. maxCn: Highest copy number to calculate for. Default is 8. male.sample: Default is FALSE. If it is a male sample put TRUE here and it will handle the XY chromosomes better. male2femref: Default is FALSE. If TRUE the sample is male but the reference you used is female. This will correct for this.To infer the arguments for patchwork.copynumbers() you will need to look at one of the chromosomal plots generated using patchwork.plot(). The structure and relationships in the plot can be interpreted to figure out the most probable locations of the allele-specific copy numbers.
For information to help you understand the axis, allelic imbalance and coverage, and layout of the plot click here. There is also a description of patchwork.plot() generated plots in Results tab.
Now lets take a look at the structure and placement of clusters on the whole genome plot. What do we expect a hypothetical plots arrangement of clusters to look like? We know that the average ploidy of the sample will be 1 on coverage axis as it is normalized. The sample may be highly rearranged but quite often this is a starting point for finding copy number 2 or copy number 3. As there will be less reads covering copy number 1 in the sample than higher copy numbers and copy number 1 cannot have different allele constitutions, by its very nature of being one allele, copy number 1 will be represented by a single cluster far to the left on coverage axis when compared to the other clusters.
Sometimes the X chromosome can be far left as well, so you may want to take a peek at the X chromosomes plot to avoid the risk of it disturbing your assessment. What if we do not have any copy number 1 in the sample? Then perhaps the far left of the plot will be occupied by two clusters, indicating the LoH and diploid state of copy number 2. It then stands to reason that the next cluster we will encounter, again; moving from left to right on coverage axis, will be copy number 3. In the same way we would expect copy number 2 to follow copy number 1 in the previous scenario.
It is with reasoning such as this, looking at the plot and how the clusters are arranged and what cluster constitutions are physically possible, that we can determine the allele-specific copy numbers in our sample.
Here is the topmost section of one such plot with some added bars and text for tutorial purposes. The plot shows the whole genome in grey and the chosen chromosome, not important in this guide, in red to blue gradient. From this whole genome picture you can then use the arrangement of the clustered areas to estimate copy numbers and allele-specific information.
The cn2 argument is the position of copy number 2. In this example cn2 is ~0.8.
The delta argument is the difference between two copy numbers on the coverage axis. In this example we used copy number 2 and copy number 3, but we could as well have used copy number 2 and the unmarked copy number 1 to the left of copy number 2. Delta does not take negative argument input. In this example delta is ~0.28.
The het argument is the position of heterozygous copy number 2 on the allelic imbalance axis. In this example het is ~0.21.
The hom argument is the position of homozygous, loss-of-heterozygosity, copy number 2 on the allelic imbalance axis. In this example hom is ~0.79.
An example run of patchwork.copynumbers():
patchwork.copynumbers(CNfile="path/to/prefix_copynumbers.Rdata",cn2=0.8, delta=0.28, het=0.21, hom=0.79)This will generate an additional set of plots in your working directory.
In the results tab we will look at interpreting the data.
Plots of patchwork.plot()
For information regarding allelic imbalance and coverage click here.
Top part of the plot
The chromosome in question is colored against a background of the complete genome in grey. A colored circles gradient and size correlate with the segments position and size on the chromosome. Colors of segments match within different fields of the plot. The circles are semi-transparent so a darker hue, both for colored and grey, indicate a greater amount of genomic content in that region.
The structure and relationships in the plot can be interpreted to figure out the most probable location of the allele-specific copy numbers. Each cluster has a certain copy number and allele content.
Quote from Execution tab:
"What do we expect a hypothetical plots arrangement of clusters to look like? We know that the average ploidy of the
sample will be 1 on coverage axis as it is normalized. The sample may be highly rearranged but quite often this is
a starting point for finding copy number 2 or copy number 3. As there will be less reads
covering copy number 1 in the sample than higher copy numbers and copy number 1 cannot have different
allele constitutions, by its very nature of being one allele, copy number 1 will be represented by a
single cluster far to the left on coverage axis when compared to the other clusters.
Sometimes the X chromosome can be far left as well, so you may want to take a peek at the X chromosomes plot to
avoid the risk of it disturbing your assessment.
What if we do not have any copy number 1 in the sample? Then perhaps the far left
of the plot will be occupied by two clusters, indicating the LoH and diploid state of copy number 2. It then stands to reason
that the next cluster we will encounter, again; moving from left to right on coverage axis, will be copy number 3. In the
same way we would expect copy number 2 to follow copy number 1 in the previous scenario.
It is with reasoning such as this, looking at the plot and how the clusters are arranged and what cluster constitutions are
physically possible, that we can determine the allele-specific copy numbers in our sample."
To the far left on the plot on the Coverage axis is a small cluster. As it has the lowest coverage in the sample and is a single cluster ,as seen in allelic imbalance context, it is reasonably copy number 1.
Moving to the right the next two clusters, lower cluster is quite small, are the allelic states of copy number 2.
Following this reasoning the next set of clusters must be copy number 3, then 4 etc. The average copy number for the sample is just above 3. This arrangement of the genome is labeled farther down in one of the plots generated by patchwork.copynumbers() or in our section above on allelic imbalance, however before using patchwork.copynumbers() you will need to be able to determine the constitution of the tumor genome as it is used as input arguments!

Middle part of the plot
Bottom part of the plot
Plots of patchwork.copynumbers()

1m0 is copy number 1, homozygous.
2m1 is copy number 2, heterozygous.
2m0 is copy number 2, homozygous.
3m1 is copy number 3, heterozygous.
3m0 is copy number 3, homozygous.
And so on.
The other chromosomal plots generated from patchwork.copynumbers() are the same as the ones generated by patchwork.plot() but with one extra part of the plot, discussed below.

In the part of the plot seen second from the top we see the total copy number, colored, and minor copy number, grey, of a segment. If a segments total copy number is 3 and minor copy number is 1 then that segment of the chromosome has 1 paternal and 2 maternal OR 2 paternal and 1 maternal copies.
Another example; if a segments total copy number is 2 and minor copy number is 0 then that segment of the chromosome has 2 paternal OR 2 maternal copies.
For more background information please contact us!
Begin by starting R. It is recommended that you use the latest version.
Enable all repositories in R so the dependencies of TAPS can be found.
setRepositories()
Install the packages that TAPS depends on to function correctly.
source("https://bioconductor.org/biocLite.R")
biocLite("DNAcopy")
biocLite("affxparser")
install.packages("fields")
install.packages("foreach")
install.packages("jpeg")
install.packages("xlsx")
#Only on unix-based systems
install.packages("doMC")
Install TAPS.
install.packages("TAPS", repos="http://R-Forge.R-project.org")
If that does not work, try installing it from source by adding 'type="source"' to the command.
install.packages("TAPS", repos="http://R-Forge.R-project.org",type="source")
If something still does not work with installation, here is a link to the location of the source file: TAPS
- Affymetrix SNP6
- Affymetrix 250K/500K
- Affymetrix CytoScan HD
Your data can be prepared using Nexus (BioDiscovery, not free), ChAS (Affymetrix, free) or manually. Using Nexus has the benefit of using their segmentation which reduces running time of TAPS.
If you have used Nexus; we recommend using the SNPRank segmentation algorithm as it is CBS based rather than HMM, then use "File -> Utilities -> Export from .ivg to.txt" to create "probes.txt", "snps.txt" and "segments.txt". These should be in a sample folder, which is what TAPS takes as input.
If you have used ChAS; load the file ending in "cyhd.cychp" into ChAS and create the text file ending in "cyhd.txt" by choosing Reports menu and selecting Export genotype results text file. Then locate the samples folder, which is what TAPS takes as input. It should now contain two files ending in "cyhd.txt" and "cyhd.cychp".
If you want to create the required input files manually, store the Log-ratio as "probes.txt" and allele frequency as "snps.txt" and place them in a folder, which will be your sample folder. These two files should have this format to be compatible with TAPS:
$less probes.txt Chromosome Start End Value chr1 15253 15278 0.2828165292739868 chr1 48168 48193 0.2047460526227951 chr1 60826 60851 -0.10802668333053589 chr1 61722 61747 0.26339590549468994 chr1 61795 61820 -0.1548314094543457 chr1 61810 61835 0.11606350541114807
$less snps.txt Chromosome Start End Value chr1 564621 564621 0.0 chr1 721290 721290 0.0 chr1 740857 740857 -5.551115123125783E-17 chr1 752566 752566 0.29908037185668945 chr1 761732 761732 1.1102230246251565E-16 chr1 765269 765269 0.0Both files are tab separated.

TAPS_plot()
The execution time of TAPS_plot() varies. Samples that have been processed by Nexus, where the segmentation step is already completed, may take anywhere from 5 minutes to half an hour. For other samples execution may take about an hour.
To run TAPS_plot(), start by initiating the R environment and loading the TAPS library:
library(TAPS)Read the documentation for TAPS_plot():
?TAPS_plotExcerpt from ?TAPS_plot regarding usage and arguments:
Usage:
TAPS_plot(directory=NULL,autoEstimate=FALSE,bin=400,cores=1,matched=FALSE)
Arguments:
directory: Default is NULL. If NULL getwd() is used. Specifying to a specific samples
directory will run TAPS_plot on that directory. Specifying a
directory containing one or more subdirectories that are
samples (and not any other subdirectories or TAPS_plot will
error when trying to run them!) will iteratively run
TAPS_plot on all samples.
autoEstimate: Default is FALSE. Development stage, do not use.
bin: Default is 400.
cores: Default is 1. Set amount of threads/cores to be used.
matched: Default is FALSE. Set to TRUE if a matched normal has been
used to remove homozygous snp allele frequencies.
The "directory" argument can be set to a folder containing several sample folders, this will execute all sample folders. For example
setting directory to the Main_folder.
Specifying "directory" to a single sample folder will execute only that sample. For example the folders Sample1 and Sample2.
Nexus

ChAS

Note that specifying any folder containing subfolders will interpret the subfolders as the sample folders.
Here is an example execution, with output, of TAPS_plot() through TAPS_call() using the current working directory.
> library(TAPS) > TAPS_plot(cores=4) [1] "Using working directory" [1] "root: /home/User/Samples/" [1] "1/4: Sample1 Loading" [1] "2/4: Sample2 Loading" [1] "3/4: Sample3 Loading" [1] "4/4: Sample4 Loading" [1] "3/4: Sample1 OK" [1] "1/4: Sample3 OK" [1] "4/4: Sample4 OK" [1] "2/4: Sample2 OK" > > TAPS_estimates() > > TAPS_click() > > TAPS_call(cores=4) [1] "1/4: Sample1 Loading" [1] "2/4: Sample2 Loading" [1] "3/4: Sample3 Loading" [1] "4/4: Sample4 Loading" [1] "3/4: Sample1 OK" [1] "1/4: Sample2 OK" [1] "4/4: Sample4 OK" [1] "2/4: Sample3 OK" >This will have generated one "<samplename>_overview.jpeg" in the main directory (should there be one), several plots and .Rdata files in the sample folder and a file named simply "SampleData.csv" which will be covered shortly.
User Evaluation
The structure and relationships in the plot can be interpreted to figure out the most probable locations of the allele-specific copy numbers. For information to help you understand the structure of the plot and how the axis allelic imbalance and average log-ratio can be interpreted, click here.
Now lets look at a general example of the structure and placement of clusters on the whole genome plot.
As average log-ratio increases, copy number increases. As Allelic imbalance increases, heterozygosity decreases, that is to say that clusters with high allelic imbalance will be homozygous.
What do we expect a hypothetical plots arrangement of clusters to look like? The average ploidy of the sample will be 0 on average log-ratio axis as it is normalized. The sample may be highly rearranged but quite often this is a starting point for finding copy number 2 or copy number 3. As there will be less reads covering copy number 1 in the sample than higher copy numbers and copy number 1 cannot have different allele constitutions, by its very nature of being one allele, copy number 1 will be represented by a single cluster far to the left on average log-ratio axis when compared to the other clusters.
What if we do not have any copy number 1 in the sample? Then perhaps the far left of the plot will be occupied by two clusters, indicating the homozygous, LOH, and heterozygous states of copy number 2. It then stands to reason that the next cluster we will encounter, again; moving from left to right on average log-ratio axis, will be copy number 3.
In the same way we would expect copy number 2 to follow copy number 1 in the previous scenario. Note also that the lowest allelic imbalance of copy number 3 MUST be higher than the lowest allelic imbalance of copy number 2 as copy number 2 can have one of each allele while copy number 3 must, at lowest allelic imbalance, have two of one allele and one of the other.
The X chromosome is shown as "X"'s instead of gradient circles and may also be far left, do not let it disturb your assessment of copy number levels as it can be misleading.
It is with reasoning such as this, looking at the plot and how the clusters are arranged and what cluster constitutions are physically possible, that we can determine the allele-specific copy numbers in our sample.
Here is the topmost section of one such plot with some added bars and text for tutorial purposes. The plot shows the whole sample in grey and the chosen chromosome, not important in this guide, in red to blue gradient. From this whole sample picture you can then use the arrangement of the clustered areas to estimate copy numbers and allele-specific information.

The cn1, cn2 and cn3 arguments are the positions of copy number 1, 2 and 3 on the "Average log-ratio" axis. In this example cn2 is ~ -0.15. You need only provide any two of these values later for TAPS_call().
The loh argument is the position of homozygous copy number 2 (LOH) on the "Allelic imbalance" axis. In this example loh is ~ 0.75.
We have developed two functions, TAPS_estimate() and TAPS_click(), that you can use if you do not want to fill SampleData.csv by hand. This is especially useful if you are running multiple samples. We would recommend that you always use TAPS_click().
TAPS_estimate()
TAPS_click()
TAPS_click() launches a GUI for sample by sample interpretation of parameters. Much like TAPS_estimate(), the output of TAPS_click() is in the form of inserting arguments for each sample into SampleData.csv. If TAPS_estimate() has been executed prior to TAPS_click(), the first thing shown will be TAPS_estimates() interpretation, see plot 1 below. At this stage you will be prompted by menu 1 and you will be given the choice to accept or redo the interpretation.
Menu 1, giving you choices to go to next sample (accept current estimates), redo the chromosome selection and interpretation of copy numbers of this sample, redo only the interpretation of copy numbers, skip the sample or close the program.
Menu 2, choose to enter an interpretation of the copy numbers or look at a specific chromosome.
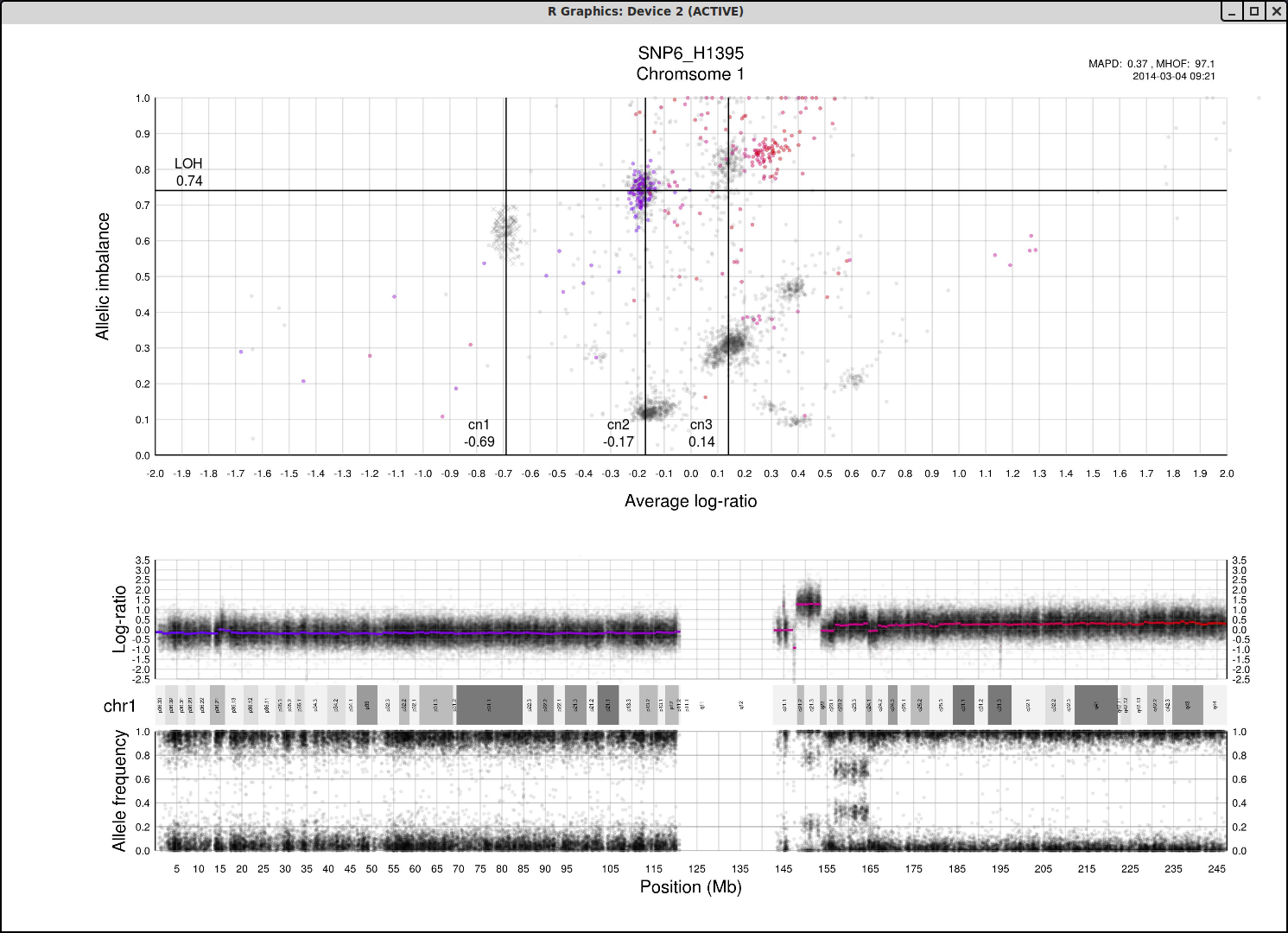
Plot 1, TAPS_estimates() estimation of parameters for a sample. In this case the estimation is correct.
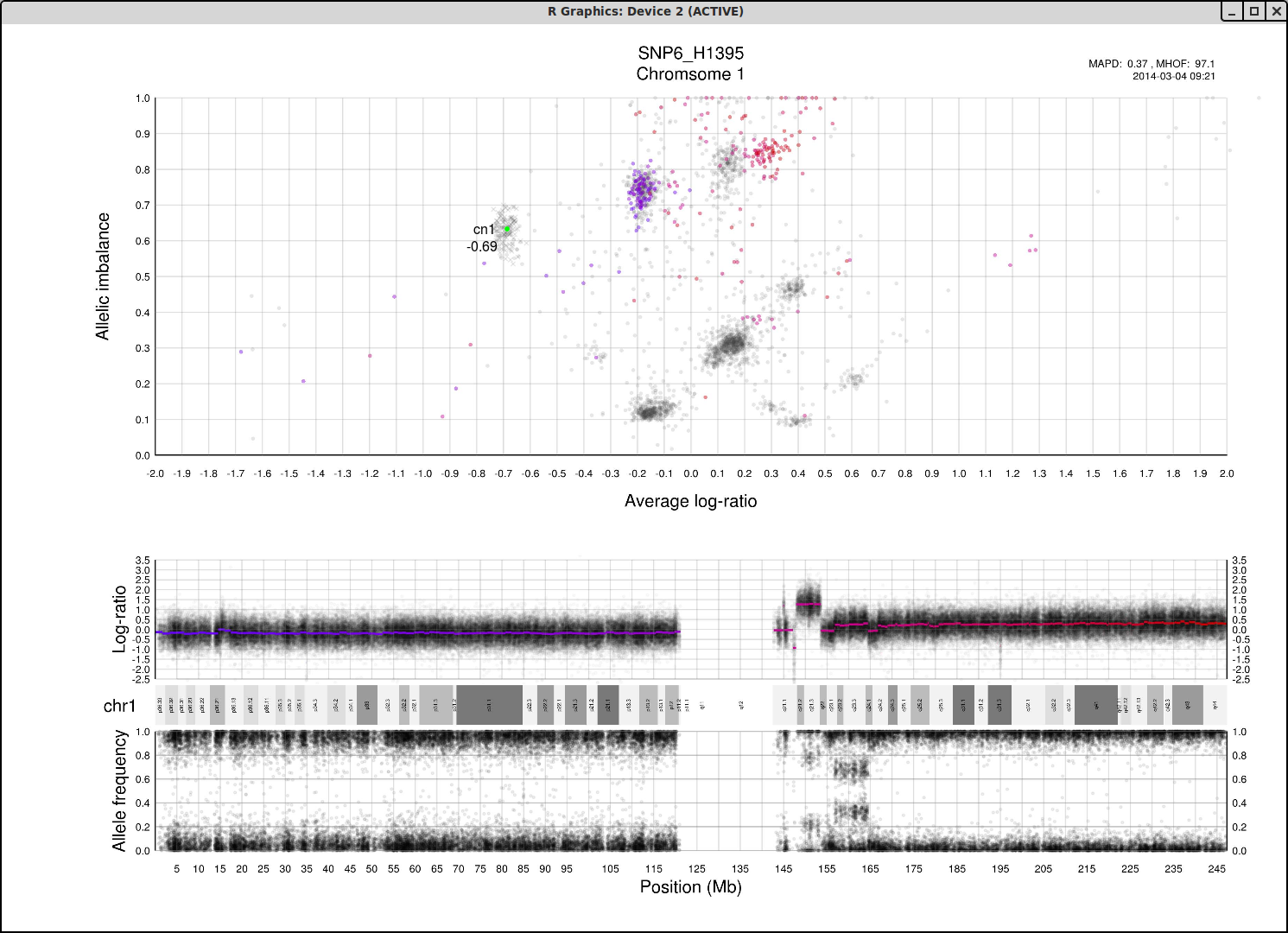
Plot 2, here we have chosen to perform copy number selection and have been prompted to identify via mouseclick the location of CN1.
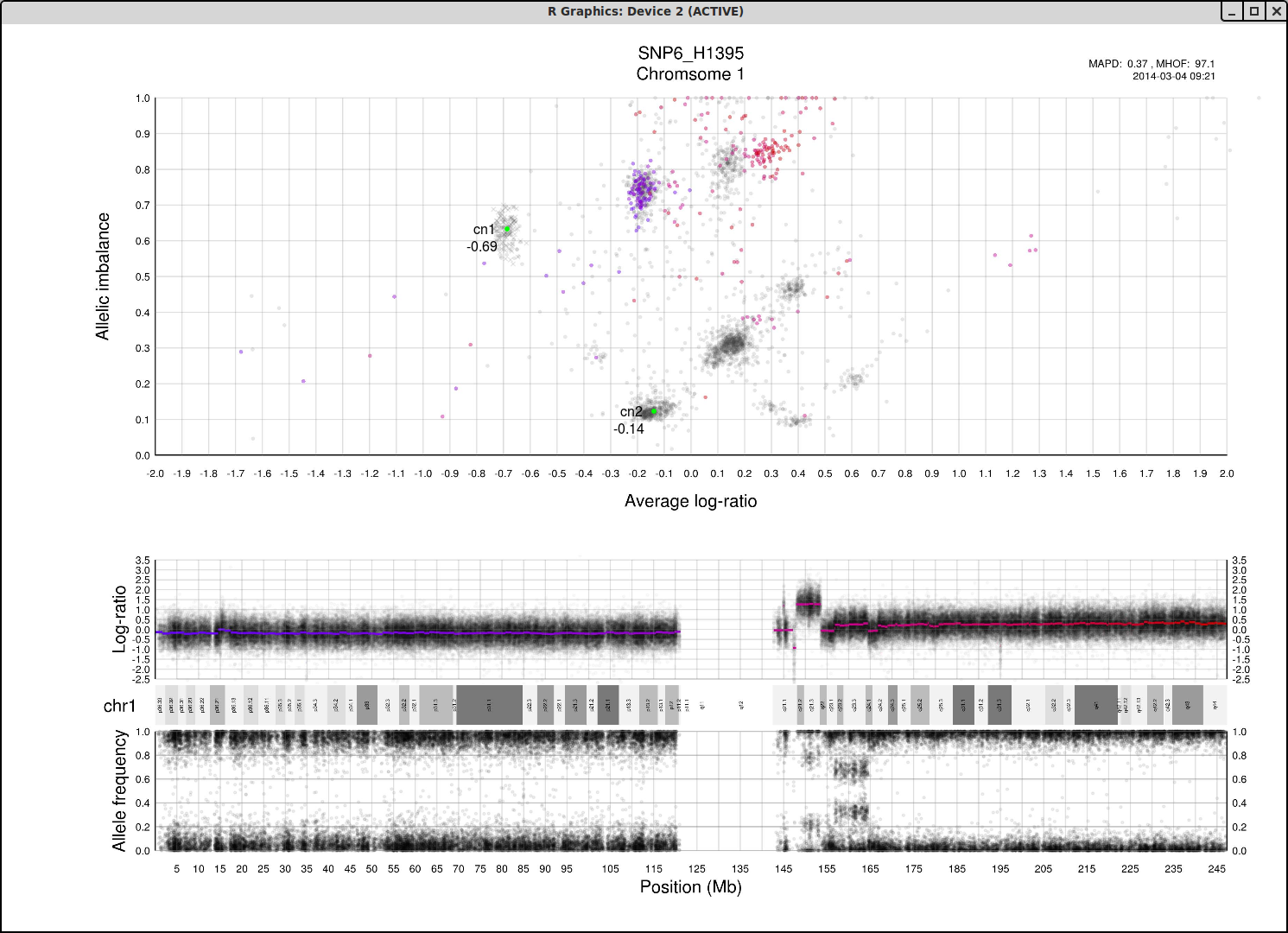
Plot 3, TAPS_click() continues to prompt for input until completion. Here we have identified CN2.
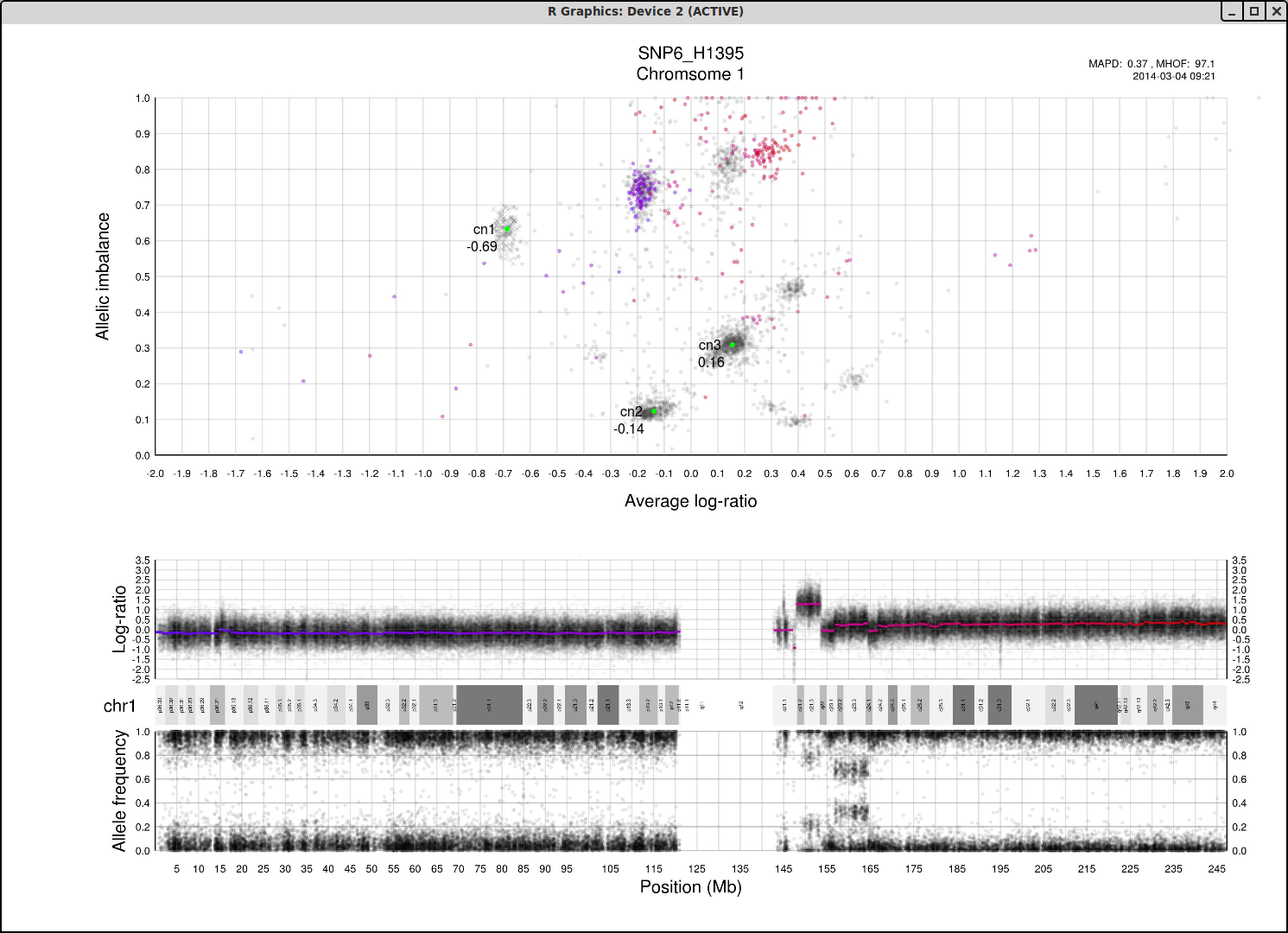
Plot 4, CN3.
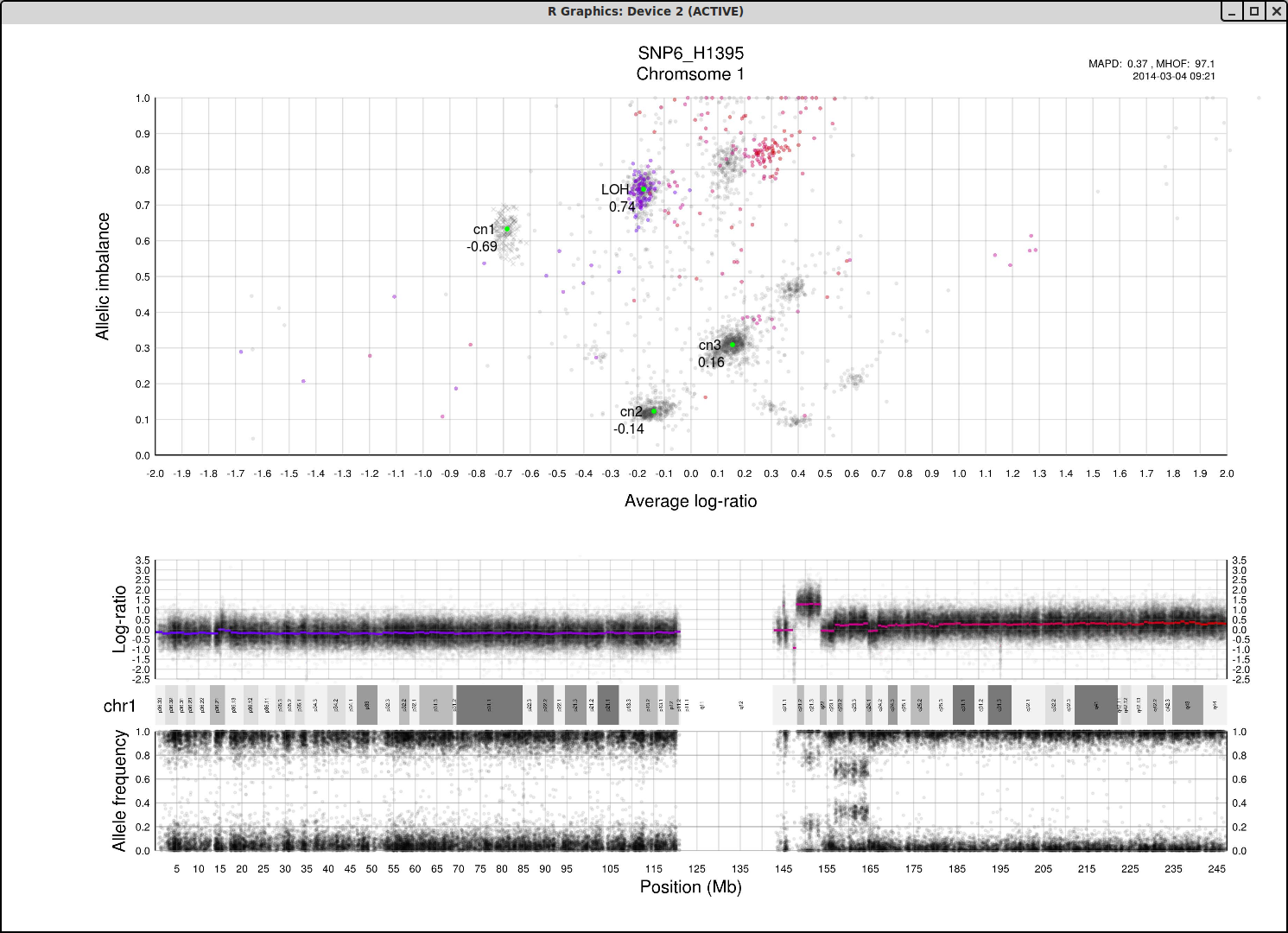
Plot 5, CN2LOH. The position of copy number 2 which has loss of heterozygosity. Located directly above copy number 2.
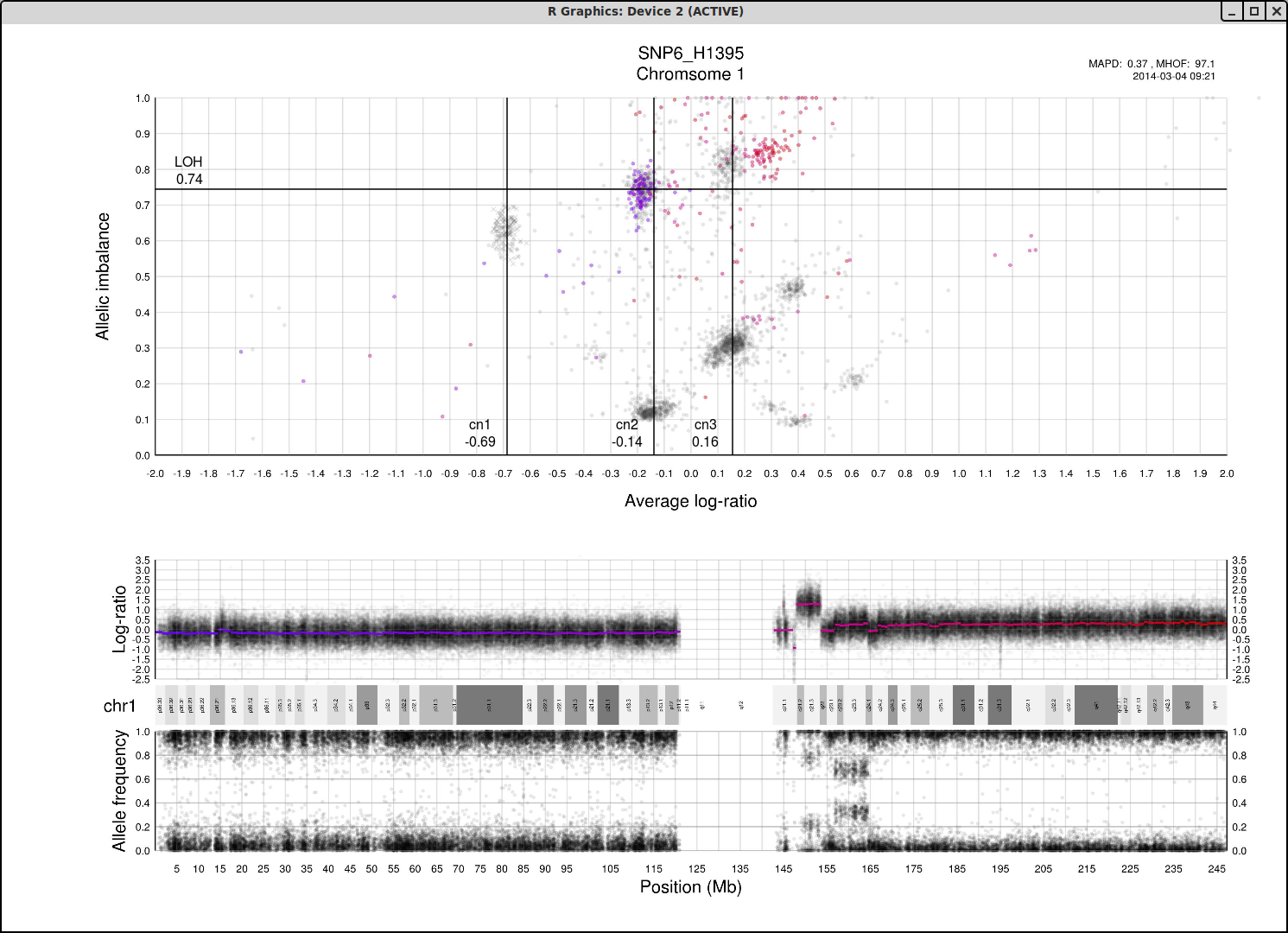
Plot 6, your new updated copy number interpretation. You will once again be prompted with menu 1 to move on to next sample etc.
TAPS_call()
TAPS_plot() must have been executed before TAPS_call() can be. This is because the .Rdata files generated during TAPS_plot() will be automatically read by TAPS_call() and you need to evaluate the plots generated by TAPS_plot() to be able to give TAPS_call() meaningful input.
TAPS_call() has "directory" as its main argument, the parameters are read from the file "SampleData.csv".
An example call to TAPS_call(), where "mainsamplefolder" contains SampleData.csv:
TAPS_call(directory="path/to/mainsamplefolder")After running TAPS_call() you will have an additional set of plots in the sample folders which includes your analysis of the plots.
In the results tab we will look at interpreting the data. If you get any errors that you can't solve, please send us an email and we will get back to you as soon as possible!
Plots of TAPS_plot()
For information regarding allelic imbalance and average log-ratio click here.
Overview plot

Top half of the plot
Bottom half of the plot
Chromosomal plots

Top half of the plot
The chromosome in question is colored against a background of the rest of the sample in grey. A colored circles gradient correlates with its segments position, seen in log-ratio, on the chromosome.
The circles are semi-transparent so a darker hue, both for colored and grey, indicate a greater amount of genomic content in that region.
The interpretation of this part of the plot especially in allele-specific copy numbers is the input for TAPS_call() and has been stated in TAPS execution tab.
Quoted here for your convenience:
"Lets take a look at the structure and placement of clusters on the whole genome plot.
As average log-ratio increases, copy numbers increases. As allelic imbalance increases, heterozygosity decreases,
that is to say that clusters with high allelic imbalance will be homozygous.
What do we expect a hypothetical plots arrangement of clusters to look like?
The average ploidy of the sample will be 0 on average log-ratio axis as it is normalized.
The sample may be highly rearranged but quite often this is a starting point for finding
copynumber 2 or copynumber 3. As there will be less reads covering copynumber 1 in the sample
than higher copynumbers and copynumber 1 cannot have different allele constitutions, by
its very nature of being one allele, copynumber 1 will be represented by a single cluster far to the left
on average log-ratio axis when compared to the other clusters.
So what if we do not have any copynumber 1 in the sample? Then perhaps the far left
of the plot will be occupied by two clusters, indicating the homozygous, LOH, and heterozygous states
of copynumber 2.
It then stands to reason that the next cluster we will encounter, again; moving from left to right
on average log-ratio axis, will be copynumber 3.
In the same way we would expect copynumber 2 to follow
copynumber 1 in the previous scenario. Note also that the lowest allelic imbalance of copynumber 3
MUST be higher than the lowest allelic imbalance of copynumber 2 as copynumber 2 can have one of each allele
while copynumber 3 must, at lowest allelic imbalance, have two of one allele and one of the other.
The X chromosome is shown as "X"'s instead of gradient circles and may also be far left, do not let it
disturb your assessment of copynumber levels as it can be misleading.
It is with reasoning such as this, looking at the plot and how the clusters are arranged and what cluster constitutions are
physically possible, that we can determine the allele-specific copynumbers in our sample."
Bottom half of the plot
Higher copy numbers will have a higher log-ratio.
Allele frequencies detected will vary depending on the allelic constitution of a segment of a chromosome. Seen here in the very frist part of the chromosome, between 0-30mb approximately, where the sample is copy number 2 with loss of heterozygosity. This means there is no variation in the allele constitution for that segment. Looking on at the rest of the chromosome in question it is copy number 3 with 2 of one allele and 1 of the other. This is shown in the banded structure, given by presence of more than one option, of allele frequency as well.
Plots of TAPS_call()
Check

This plot, arbitrarily named "check", is not chromosome specific and shows the complete tumor sample with assigned tags for copy number and allele content, much like the explanatory picture seen previously but with text describing the copy numbers and allele ratios of the segments rather than purple and green bars.
1m0 is copy number 1, homozygous.
2m1 is copy number 2, heterozygous.
2m0 is copy number 2, homozygous.
3m1 is copy number 3, heterozygous.
3m0 is copy number 3, homozygous.
And so on.
If the texts showing the approximate positions of the allele-specific copy numbers seem missplaced you may want to try to improve, or tweak, your input values in SampleData.txt, the parameters used by TAPS_call().
Chromosomal plots with total and minor copynumbers

Left half of the plot
Right half of the plot
Another example; if a segments total copy number is 2 and minor copy number is 0 then that segment of the chromosome has 2 paternal OR 2 maternal copies.
Below that we see the same three plots of log-ratio, cytoband information and allele frequency as seen above in chromosomal TAPS_plot() plots. Notice that all four of the plots on the right side have the position on the chromsome in question as their x axis.
TAPS_Region()
Usage:
TAPS_region(directory=NULL,chr,region,hg18=F)
Arguments:
directory: Default is getwd().Directory must contain TAPS_plot_output.Rdata,
an output from TAPS_plot(), which contains all information that TAPS_region() needs.
chr: Supply the chromosome you wish to look at. ex: chr=1
region: The region of the chromosome you are interested in. ex:
region=10000000:13000000
hg18: Default is FALSE, meaning the hg19 known gene list will be
used. If TRUE the hg18 known gene list is used.
Now lets take a look at an example plot of region 18000000-23000000 of chromosome 1 for sample 6295.
TAPS_region(chr=1,region=18000000:23000000)

Most of the content of the plot has been discussed above. Unique to the TAPS_region() output is the red "Selected region" marker on the whole chromosome view, which spans logRatio, cytoband information and allele frequency. This selected region is then shown in the bottom half of the plot, with an additional row of known gene information.
Notice that you chose the size of the region. Larger regions will have gene information that is hard to discern whereas very small regions might seem void of content. About 5 million basepairs, as seen here, or smaller is recommended.
For more background information please contact us!
Changelog
Here you can find the recent changes made to the patchwork and patchworkCG packages.TAPS 2.1 -
04/03/2014
- New function: TAPS_estimate(). Automatic estimation of allele-specific copynumbers for all samples that have completed TAPS_plot()
- New function: TAPS_click(). Use a GUI to accept TAPS_estimates() values or add your own interpretation through direct interaction with the TAPS_plot() plots.
- New function: TAPS_region(). Zoom in on a region specified by user which includes known gene information.
- Updates to homepage regarding TAPS usage.
patchwork v2.4 & TAPS 2.0 -
11/09/2013
- Complete makeover of patchwork plots visualization. Now includes cytoband information.
- New function: patchwork.region(). Shows a region you specify of a chromosome. Contains Known Gene information
- New function: TAPS_compare(). Compare aberration frequencies in two groups of samples.
- New function: TAPS_freq(). Summary of aberration frequencies in several samples.
patchwork v2.3 -
16/08/2013
- Moved Pysam out of package due to auto-install issues. Now you will have to manually install it. See Patchwork -> Installation
patchwork & TAPS -
18/06/2013
- Buggfix for Patchwork
- Buggfixes for TAPS
- Added information regarding the tool TAPS to homepage
- Patchwork now gives you an additional file, <sample_name>_somatic.Rdata, which contains possible, unfiltered, somatic variants.
Homepage -
11/09/2012
- Changes and updates to all information in tabs under Patchwork
patchwork v2.2 -
31/08/2012
- Parameter naming changes and updates
- Implemented mpileup compatability so that latest versions of SAMtools can be used. Note that it is still more efficient to use old pileup command
- Fixed an issue we were seeing with GC normalization when HG18 was used
- Fixed an issue with some plots being out of bounds when HG18 was used
patchwork v2.1 -
25/06/2012
- Improved sex-chromosome handling
- Added support and documentation for HG18
- Updated naming of files created from running patchwork
patchworkCG v2.0 -
17/04/2012
- Improved segmentation for Allelic Imbalance
- Changes to onLoad message handling
No older changes have been recorded.